The purpose of DeepSeq2Drug is to screen potential drug for novel viruses virtually. Some virus-drug datasets make using machine learning-based methods for drug repurposing possible. However, those datasets only provide known pairs, while few provide other features/embeddings. Thus, the most recently proposed methods are network-based (graph-based) methods. The advantages of the graph-based method are making the most of prior known pair knowledge and searching for potential links between viruses and drugs. Nonetheless, graph-based methods are limited when coming across novel viruses, as if there are no known links between novel viruses and drugs. Also, the mutated virus can not be dealt with using the graph-based method because all these mutants are regarded as the same virus in the graph.
The DNA and RNA contain undiscovered information. And for each virus mutant, the sequence varied a lot, especially for the RNA virus. Also, for viral sequences, there is a database (NCBI virus [1]) that can provide sequences for each mutant, making collecting sequences for each virus possible.
Thus, using viral sequence features to model drug repurposing is promising, more feasible, and flexible.
Many aspects of viral features can contribute to the virtual drug screen,
Such as sequence motifs (patterns), structures, statistical features, and so on. Based on our previous work [2], we started with statistical calculation features (which are calculated from the ilearn platform[3]). Based on another previous work[4], we also introduced an NLP-based method, doc2vec[5], to extract latent embedding based on virus sequences.
In the next version of DeepSeq2Drug, we might introduce sequence motifs and RNA structures (2D or 3D) to polish our prediction system further.
To further explore the mutant RNA viruses, we took different mutants of the sars-cov-2 virus and conducted more experiments.
We first applied MAFFT[6] to five (four mutants+one reference sequence ) sars-cov-2 virus mutants, including B1, BA5, BF7, and XBB.1, and constructed a phylogenetic tree for these mutants (Shown in Figure 1). The B1 is the early mutation of sars-cov-2, while BA5, BF7, and XBB.1 are the later ones.
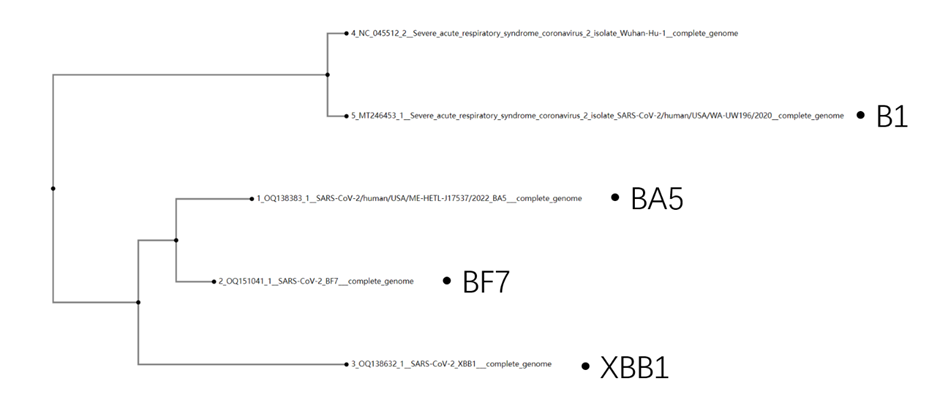
Figure 1, the phylogenetic tree of sars-cov-2 virus mutants.
We first compared Pearson Similarity (0-1, close to 1 means the vectors are very similar) based on the doc2vec embeddings of each mutant. Results are shown in the following:
B1,BA5 PearsonRResult(statistic=0.9006671139755364, pvalue=1.9208206333532863e-47)
B1,BF7 PearsonRResult(statistic=0.9019241075574765, pvalue=8.964827874717818e-48)
B1,XBB1 PearsonRResult(statistic=0.9067403418932873, pvalue=4.382863371575376e-49)
BA5,BF7 PearsonRResult(statistic=0.9983050803791067, pvalue=1.697090393959078e-157)
XBB1,BA5 PearsonRResult(statistic=0.9981705601918953, pvalue=2.077632850541821e-155)
XBB1,BF7 PearsonRResult(statistic=0.9976816181777338, pvalue=6.190036926014514e-149)
BA5, BF7, and XBB.1 are the mutants that appeared closely in time. Thus, as can be seen from the similarity, these three mutants might be similar (around Pearson 0.99). BA5 and BF7 have the highest correlation value of 0.9983, which aligns with the phylogenetic tree. All of the later mutants BA5, BF7, and XBB.1 have less Pearson similarity with their ancestor mutant B1 (almost around 0.9).
To explore how the mutation might influence the prediction results, we introduced the rank correlation coefficient. The Kendall rank correlation coefficient evaluates the degree of similarity between two sets of ranks given to the same set of objects. If the Kendall rank correlation is close to 1, the two predictive ranks of the mutants are almost the same. Thus, the Kendall rank correlation can reflect the rank variance among mutants.
To better analyze the Kendall rank correlation variance
We selected three separated predictors (doc2vec-resnet50, doc2vec-GPT, and doc2vec-role2vec) plus ensemble results. We compared BA5, BF7, and XBB.1 with B1 for the top 20, top 30, top 50, top 70, and top 100 results predicted by DeepSeq2drug software.
The results of the Kendall rank correlation variance are shown in Figure 2-5.
For different predictors, the Kendall rank correlation can vary differently.
As can be seen from Figure 2 to Figure 4, the Kendall correlation varied a lot for different predictors (vdkey) and mutants. For doc2vec-Resnet50, BA5 and BF7 are more likely to be the same (similar to the phylogenetic tree), while for the other two predictors, BF7 and XBB.1 are more likely to be the same (in rank). One thing in common is that the top 20 and 30 have an overall higher Kendall rank correlation for the separate predictor. Figure 5 demonstrates the Ensemble results in the Kendall correlation comparison of BA5, BF7, and XBB.1. For the ensembled result, the top 20 to top 30 Kendall correlation becomes much lower. This might indicate that the ensemble strategy can give the same group of mutants more varied top results, helping the algorithms screen varied drugs for different mutants better. For the Top 50, 70, and 100 results, it is reasonable to have a higher Kendall correlation as these mutants still belong to the same virus group.
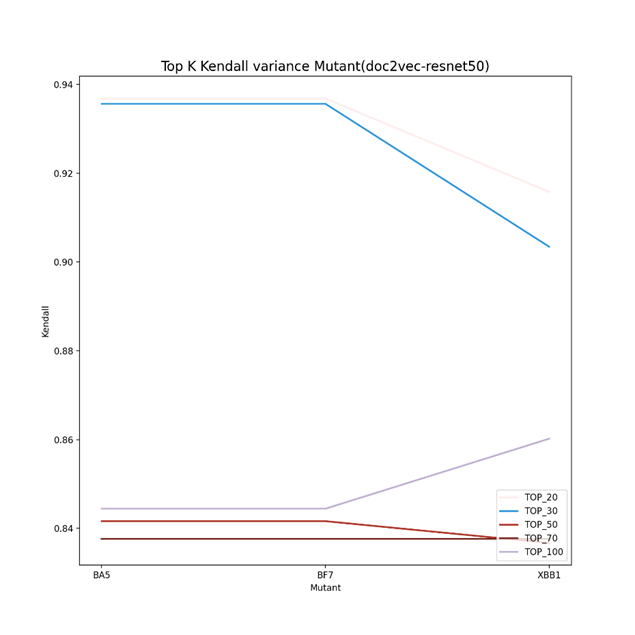
Figure 2. Top K Kendall correlation using the vdkey of doc2vec Resnet50(K=20,30,50,70,100). The Kendall correlation varied a lot for different predictors (vdkey) and mutants. BA5 and BF7 are more likely to be the same (similar to the phylogenetic tree)
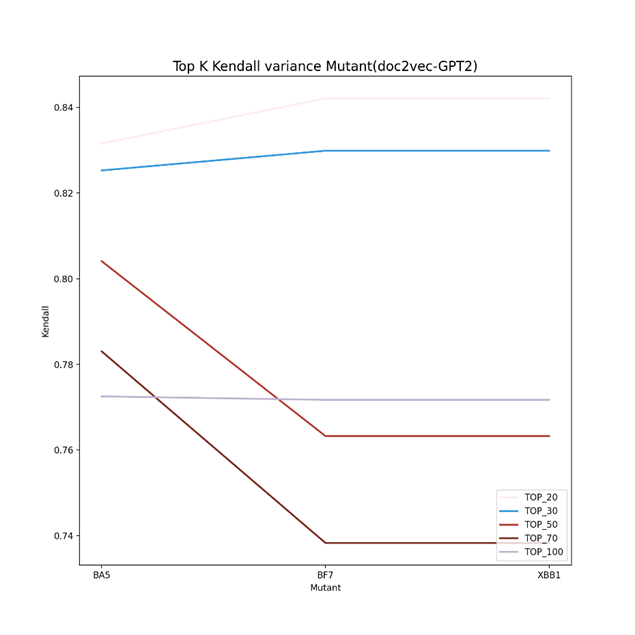
Figure 3. Top K Kendall correlation using the vdkey of doc2vec GPT2(K=20,30,50,70,100). The Kendall correlation varied a lot for different predictors (vdkey) and mutants. BF7 and XBB.1 are more likely to be the same (in rank)
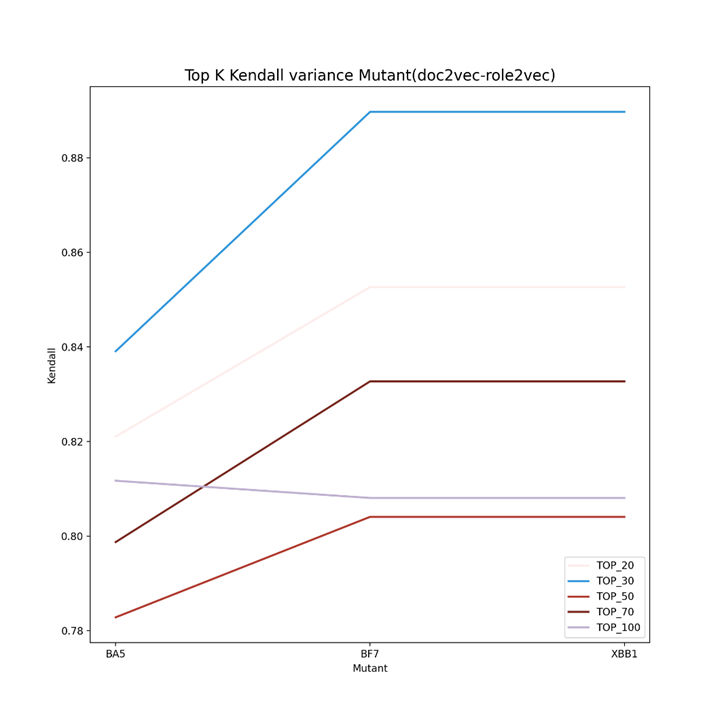
Figure 4. Top K Kendall correlation using the vdkey of doc2vec-role2vec (K=20,30,50,70,100). The Kendall correlation varied a lot for different predictors (vdkey) and mutants. BF7 and XBB.1 are more likely to be the same (in rank)
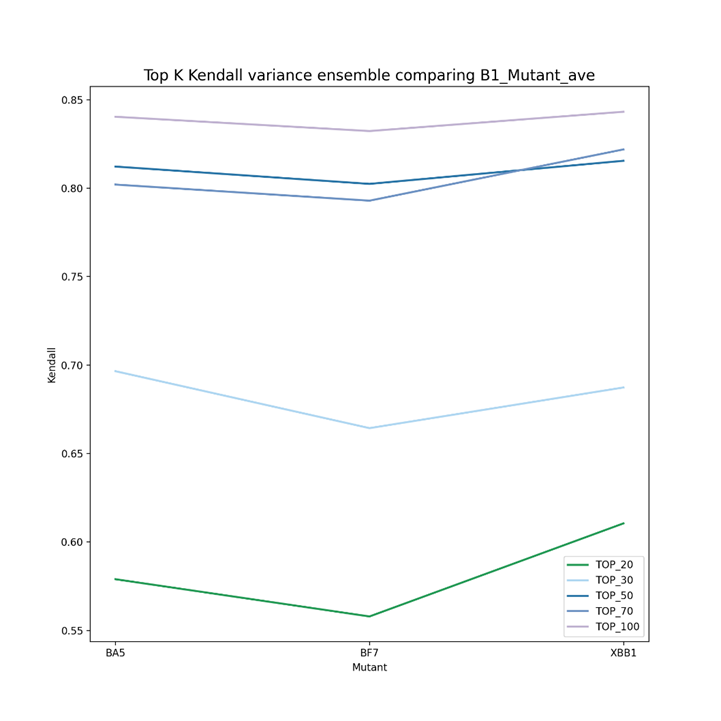
Figure 5. Ensemble results in Kendall correlation comparison of BA5, BF7, and XBB.1 with B1. For the ensembled result, the top 20 to top 30 Kendall correlation becomes much lower, indicating that the ensemble strategy can give the same group of mutants with more varied top results. For the Top100 results, it is reasonable to have a higher Kendall correlation as these mutants still belong to the same virus group.
Case Study
One research group [7] showed that Omicron variants (BA5) have a higher sensitivity to inhibition by soluble angiotensin-converting enzyme 2 (ACE2) and the endosomal inhibitor chloroquine compared to D614G (B1).
Take the Rank of Chloroquine as an example, as shown in Table 1. We found that for both the sum results and the averaged results predicted by DeepSeq2Drug, the rank of chloroquine is higher for Omicron variants (BA5), which is in alignment with the previous study [7], indicating the potential predictive ability of DeepSeq2Drug to not only virus but also its variant.
Table 1. The rank of chloroquine predicted with different mutants
| Mutants | Rank of chloroquine (Sum) | Rank of chloroquine (Ave) |
| BA5 Omicron variants | 13 (higher sensitivity) | 2 (higher sensitivity) |
| B1 (D614G) | 19 | 4 |
| BF7 | 19 | 4 |
| XBB.1 | 17 | 4 |
Summary
In summary, by using Pearson Similarity and Kendall rank correlation coefficient, we evaluate that the proposed model can predict mutants from the same group of viruses with varied drug results. A case study of the Rank of Chloroquine further indicates the potential predictive ability of DeepSeq2Drug to the virus and its variant.
Reference
- Brister J R, Ako-Adjei D, Bao Y, et al. NCBI viral genomes resource[J]. Nucleic acids research, 2015, 43(D1): D571-D577.
- Xie W, Chen X, Zheng Z, et al. LncRNA-Top: Controlled deep learning approaches for lncRNA gene regulatory relationship annotations across different platforms[J]. Iscience, 2023, 26(11).
- Chen Z, Zhao P, Li F, et al. iLearn: an integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data[J]. Briefings in bioinformatics, 2020, 21(3): 1047-1057.
- Xie W, Luo J, Pan C, et al. SG-LSTM-FRAME: A computational frame using sequence and geometrical information via LSTM to predict miRNA–gene associations[J]. Briefings in bioinformatics, 2021, 22(2): 2032-2042.
- Le Q, Mikolov T. Distributed representations of sentences and documents[C]//International conference on machine learning. PMLR, 2014: 1188-1196.
- Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program[J]. Briefings in bioinformatics, 2008, 9(4): 286-298.
- Neerukonda SN, Wang R, Vassell R, Baha H, Lusvarghi S, Liu S, Wang T, Weiss CD, Wang W. 2022. Characterization of Entry Pathways, Species-Specific Angiotensin-Converting Enzyme 2 Residues Determining Entry, and Antibody Neutralization Evasion of Omicron BA.1, BA.1.1, BA.2, and BA.3 Variants. J Virol 96:e01140-22.https://doi.org/10.1128/jvi.01140-22