An imbalanced dataset might have an impact on AUPR. The following experiments show more details about how the imbalance could contribute to AUPR.
- First, our final predictors are trained in the balanced dataset. Thus, as long as one pair of vdkey performed the best (Significantly higher AUC+AUPR value in their feature-view domain) in our feature pool experiments, this pair of vdkey has the best predictive ability to predict potential pairs of viruses and drugs under the balanced scenario. The dataset construction and feature selection are separate parts of our designed experiments.
- Second, the imbalance issue can be an interesting topic to explore, as it can reflect the potential robustness of the proposed method. Here, we introduced a Negative sampling Rate (NR) as our previous study [1] did and conducted more experiments on this issue. In the situation of a balanced dataset, the NR equals 1, and the larger the NR is, the more the negative samples were sampled during the phase of dataset construction.
More details of the experiments are disclosed below:
NR experiments details:
Hardware Platform:
GTX4080 Laptop GPU, Intel i9-13900HX, 64GB DDR5 memory
Vdkey selection:
We selected seq-embedding, doc2vec, and corpus-embedding, Albert for the virus, and GPT2, bioGPT, role2vec, and resnet50 for the drugs.
NR range:
(0.5,1,2,4,8,16)
Dataset:
DrugVirus2.0, five-fold cross-validation for each NR, we collect the mean value of AUC/AUPR/Metric and draw a curve line chats for analysis.
Results
Figures 1-2 are the curve charts for fixed virus embeddings. Figures 3-5 are the curve charts for fixed drug embeddings. Figures 6-7 are the curve chats for the extra experiments embeddings (the expanded embeddings).
The X-axis is the Negative sampling Rate (NR), while the y-axis is the AUC, AUPR, and metric from the top to the bottom. Different color denotes different vdkeys. Different line types indicate AUC/AUPR/Metric.
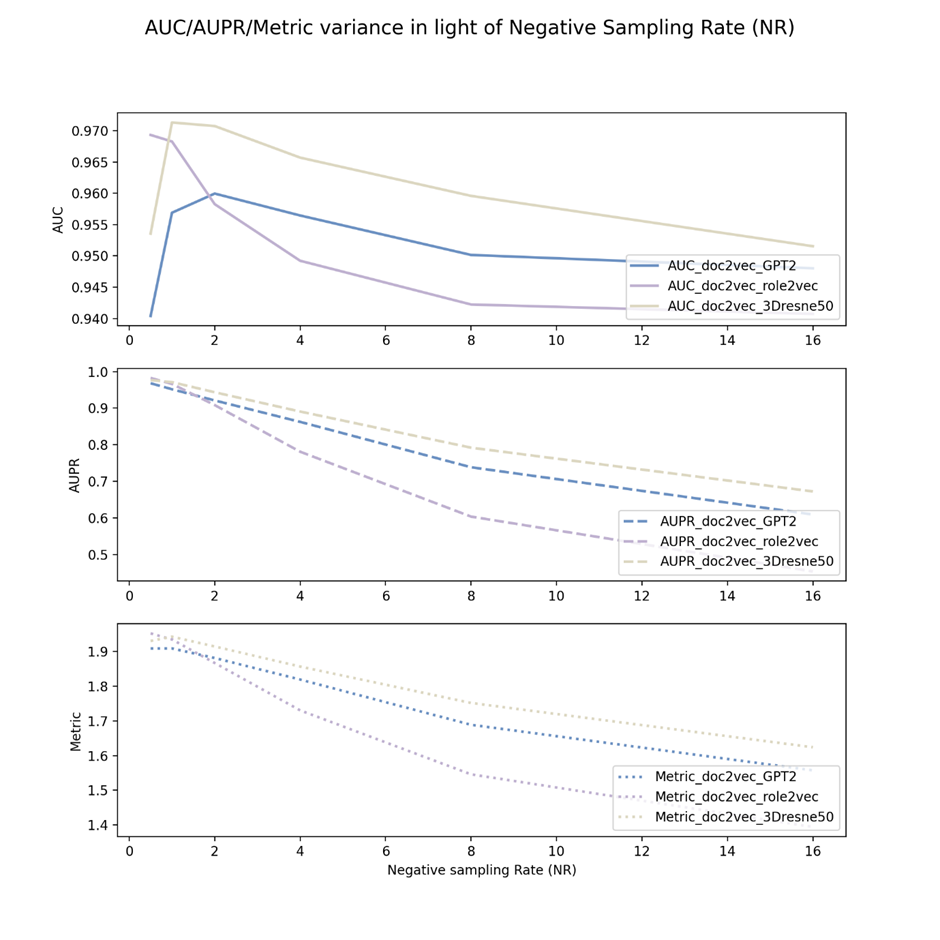
Figure 1 shows the negative sampling rate experiments of doc2vec viral embeddings and three drug keys from different feature view domains.
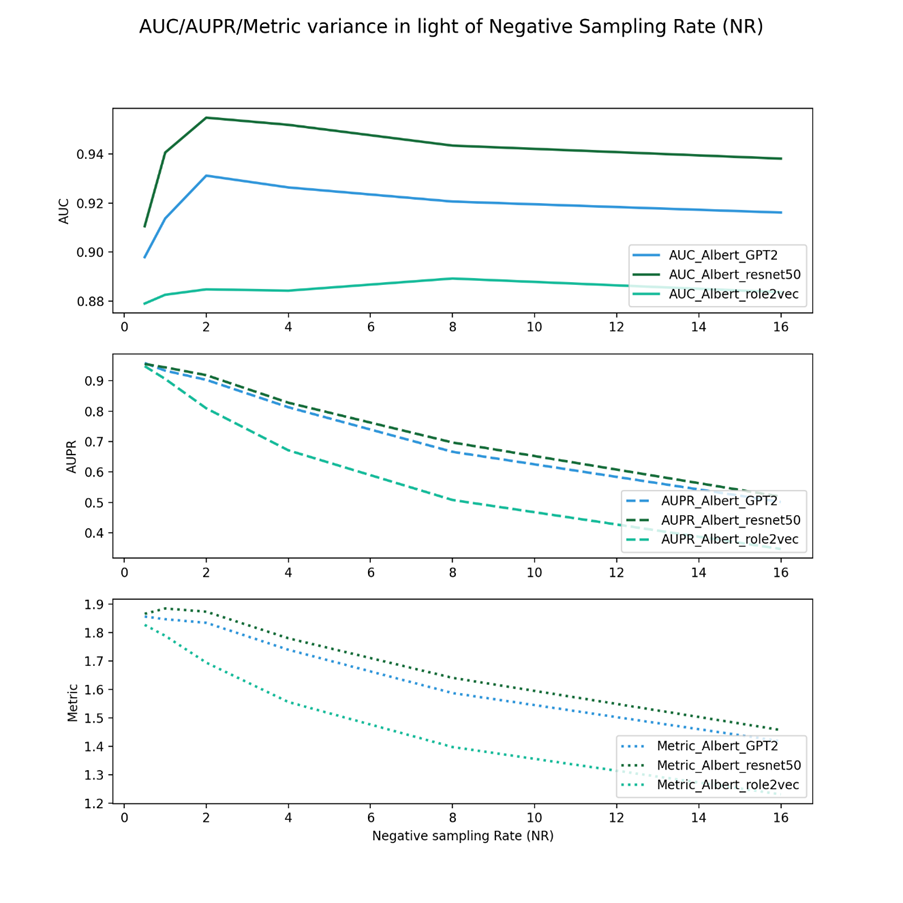
Figure 2 shows the negative sampling rate experiments of Albert viral embeddings and three drug keys from different feature view domains.
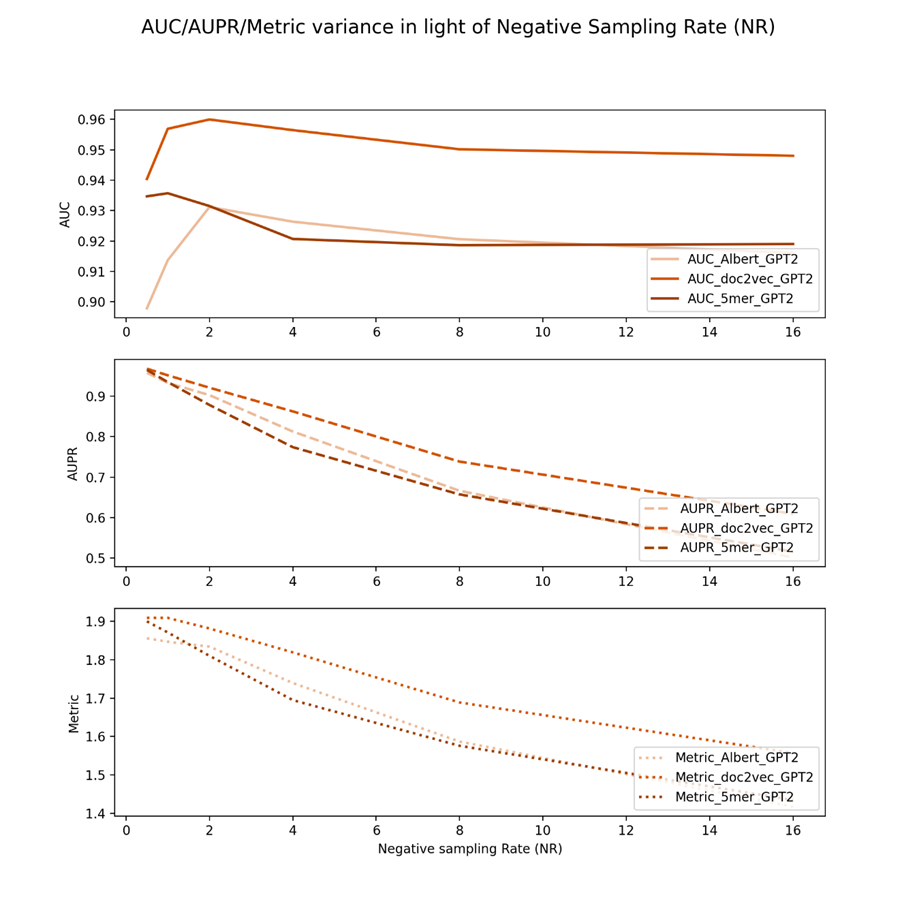
Figure 3 shows the negative sampling rate experiments of drug GPT2 embeddings and three virus keys from different feature view domains.
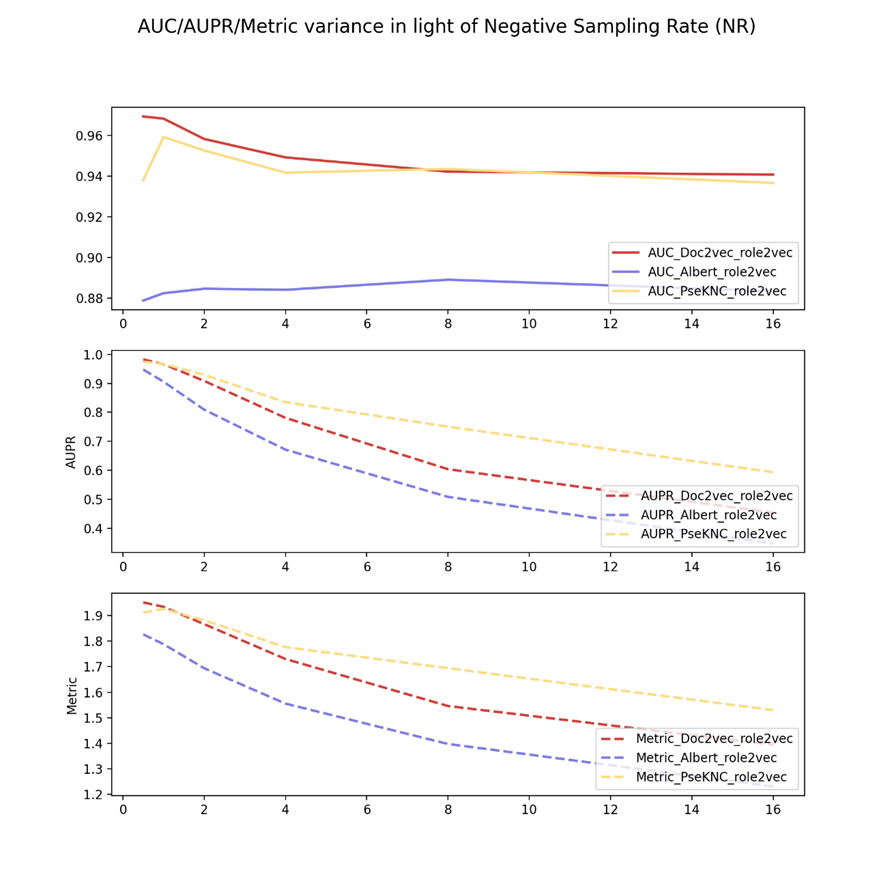
Figure 4 shows the negative sampling rate experiments of drug role2vec embeddings and three virus keys from different feature view domains.

Figure 5 shows the negative sampling rate experiments of drug 3Dresnet embeddings and three virus keys from different feature view domains.
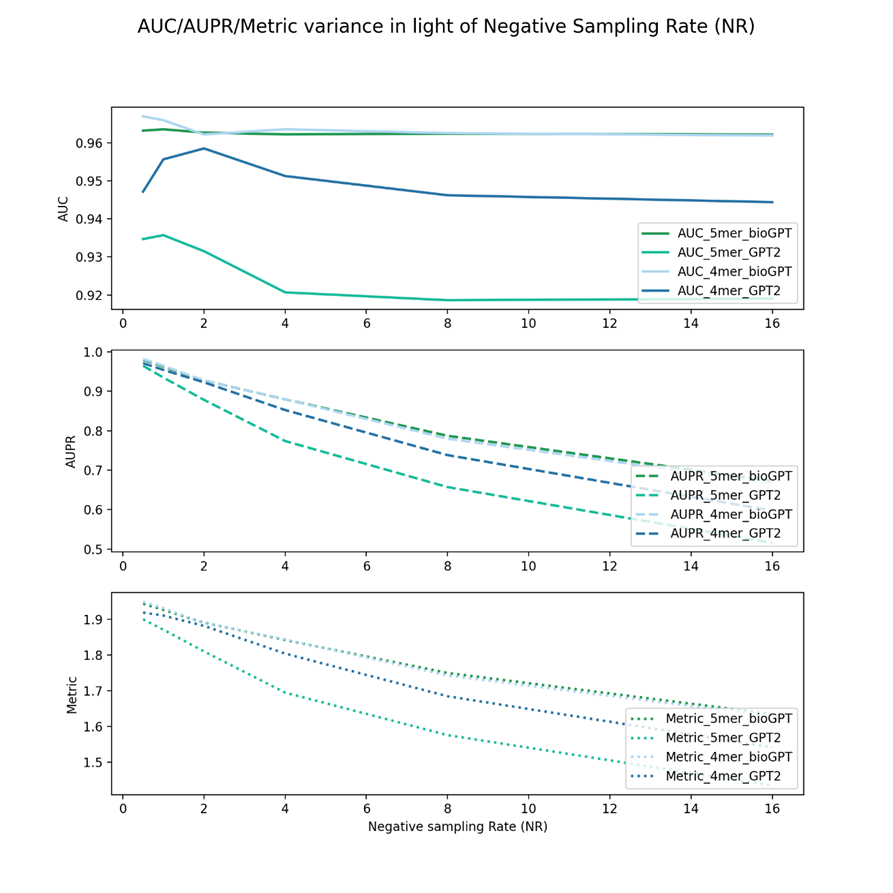
Figure 6 shows the negative sampling rate experiments of drug GPT2 and bioGPT embeddings and two virus sequence feature keys, 4mer and 5mer. As can be seen, bioGPT can achieve better performance in AUC, AUPR, and metric.
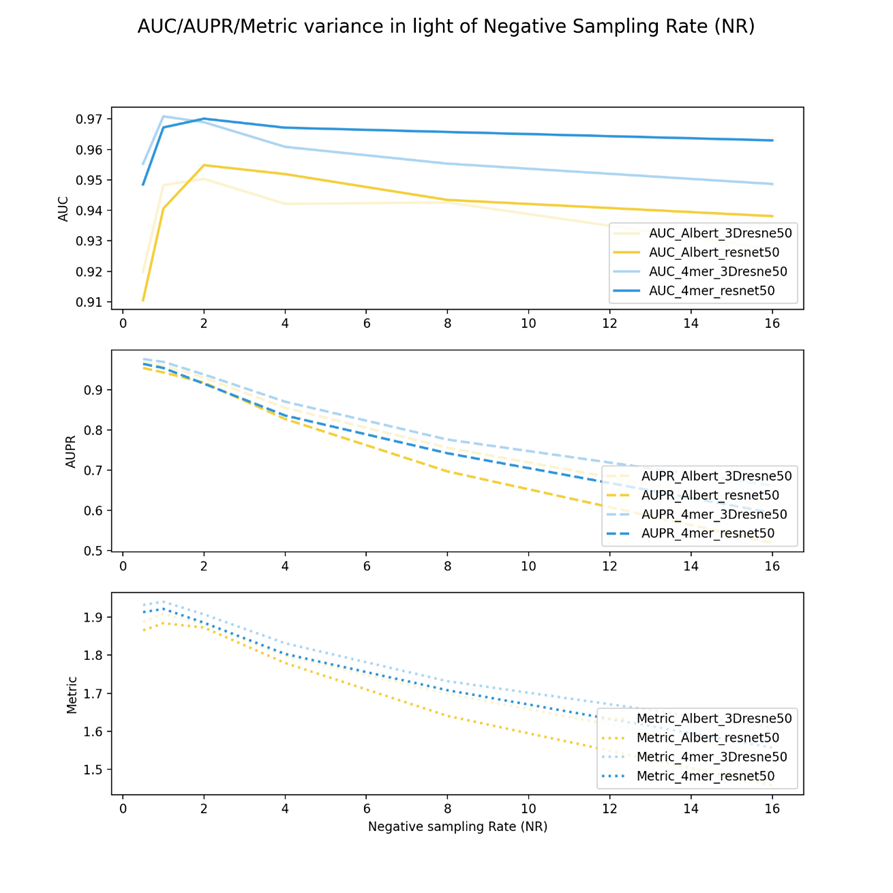
Figure 7 shows the negative sampling rate experiments of drug resnet50 and 3Dresnet50 embeddings and two virus sequence features or corpus embedding keys, 4mer and Albert. For the AUC, after the NR more than 1, resnet50 seems to behave better. Nonetheless, from the metric/AUPR aspect, the 3D resnet50 can perform better than the resnet50 among all NR cases. This indicates that the increment of AUPR is more than AUC, as metrics have the same trend with AUPR.
Summary
In summary, as can be seen from all these results, in most cases, AUC values decreased but not on a large scale. Especially for Figures 2,3,4,6, as NR increased, the AUC almost didn’t decrease a lot. In contrast, AUPR varied a lot in alignment with NR. When the NR increased, for all of the vdkeys, AUPR decreased rapidly.
This might be because the model we leveraged is only a machine-learning model with an ensemble method; the machine-learning RF model is not that robust if the dataset becomes very imbalanced. However, the fact is that the known pairs for virus-drug are far more sparse than our previous work [2]. In that case, micro-RNA and genes have more than 500,000 pairs, and the numbers are still increasing. For our virus-drug pairs, we only have less than 1500 pairs of positive samples, and if we introduce many more negative samples, the predictor might not be able to distinguish positive samples as there are not enough numbers. Using a deep learning model might be another option to increase the proposed ensemble model’s robustness, as seen in our last paper[1]. However, as the dataset is relatively too small, using DNN models might inevitably face overfitting challenges. We might explore those experiments in the next generation of DeepSeq2drug.
Another trend we can find is that for most of the cases (except for figures 1 and 4), the metric values are more related to vdkeys. If the vdkeys are fixed, no matter how the negative sample numbers change, the rank of the metric remains the same, which indicates that the metric might be a good indicator for selecting the vdkeys by feature pool.
From Figure 6 and Figure 7, we can further know that those who have gained higher metrics in the expanded experiments also performed better in the NR experiments. This indicates that the quality of the embedding might have an important role in model performance. Thus, we will pay attention to more advanced models/algorithms and use those methods in our framework.
Reference
- Xie W, Chen X, Zheng Z, et al. LncRNA-Top: Controlled deep learning approaches for lncRNA gene regulatory relationship annotations across different platforms[J]. Iscience, 2023, 26(11).
- Xie W, Luo J, Pan C, et al. SG-LSTM-FRAME: A computational frame using sequence and geometrical information via LSTM to predict miRNA–gene associations[J]. Briefings in bioinformatics, 2021, 22(2): 2032-2042.