DeepSeq2Drug: An Expandable Ensemble End-to-end Antiviral Drug Repurposing Benchmark Framework by Multi-Modal Embeddings and Transfer Learning.
Extra Experiments
EXP 1 Time-complexity/ Runing time
EXP 2 comparison-with-sota-methods
EXP 3 similarity-of-sequences-in-the-ctd-dataset
EXP 6 Corpus-embedding-contribute-to-the-predictive-results
Supplementary Materials Contents
More details concerning the pre-trained models can be found in Supplementary Materials. (PART I Features/embedding)
Part II Datasets
Details about the negative sampling strategies are shown in Supplementary Table 3.
Part Ⅲ Results
The visualized results (violin-swam plot), dataset overlap, and the p-value matrix of transfer results can be found in Supplementary Materials, Part III.
Part Ⅳ Extended Results and Parametrical Analysis
Part V Case Study of RSV
Additionally, we checked the findings for the respiratory syndrome virus (RSV); 20 out of the top 20 predictive results were reported (further information regarding the RSV prediction outcomes can be found in Supplementary Table 5.
—————————————————————————————–
PART Ⅰ Features/embedding
This manuscript mainly focused on embeddings from sequences, corpus, images, and networks. For viruses, the DNA sequence and virus-related abstracts/descriptions are more accessible. Thus, we mainly leveraged sequence-based and corpus-based (NLP) embeddings (for the virus). Only a few drugs have sequences. We didn’t consider drug sequences in this framework to maximize the number of drugs that can generate all modal feature embedding. We leveraged descriptions, molecular images, and networks to generate drug features/embeddings. We also introduced a virus or drugs feature from GCNMDA [1], downloaded from its official GitHub, to compare with our methods in embedding quality. The sources of previous contents and candidate methods leveraged for extracting features/embeddings are shown in Supplementary Table 1.
Supplementary Table 1. Feature/embedding types and candidate extractors
| Feature-view domains | Source | Candidate Virus features/embedding extractors | Candidate Drug embedding extractors |
| Sequence-based ( iLearn [2]) | NCBI virus DNA sequences [3] | 4mer,5mer, mismatch41, PseKNC2013, Z48bit | N/A |
| Sequence-based embedding (Doc2vec) | NCBI virus RNA sequences | Doc2vec (model trained from 867020 sequences) | N/A |
| corpus-based (NLP) embedding (7984) | PubMed Collected Virus-related Papers abstracts | Roberta/ Albert/Bert (finetune selected Roberta and Albert)* | Bert/Gpt-2/Roberta/ Albert/ (Sources: drug-bank descriptions [4]) |
| Molecular Image-based Embedding (9212) | DrugBank Molecular Image [4] | N/A (9212) + | Resnet50, Resnest101, Rfficient net, Inception-Resnet |
| Networks-based Embedding | Drug-Drug-interaction, Drug-Target interactions [4], Drug-disease interactions | N/A | Role2vec, node2vec, GCNMDA (SOTA method) (finetune selected Role2vec 768 and Role2vec 768(30) ) |
* Gpt-2 is not able to be applied to virus abstract corpus due to the limitation of the GPU memory of the experiment platform. The Platform is i7-9700 CPU, 64 Gigabytes of Memory, and GTX2080Ti, 11 Gigabytes of graphic memory. Each feature-view domain may contain more than one feature extractor.
Part II Datasets
Supplementary Table 2. Details of the collected datasets/databases
| Database | No. virus | No.drugs | No.interactions | Type | purpose |
| NCBI virus [3] | Expandable | Not applicable | N/A | Virus seq | Virus-sequence based features |
| DrugBank [4] | N/A | 190 (13,473) | N/A | Drug info | Drug features (corpus, Image, network) |
| Drugvirus [5] | 103 (153) | 190 (231) | 1,156 (1,518) | Drug-virus Relation | Constructed datasets Train model |
| CTD [6]. | 11 (2 overlap) | 585 | 1,588 | Chemical-virus relations | Transfer Test |
| DrugBank: Drug-Target [4] | 4,596 (seq) | 7,381 (Overlap with DrugVirus:136) | 20,127 | Drug-target relations | Network construction |
| Drug-Drug-interaction, | N/A | 391 (ODV: 56) | 875 | Drug-Drug Interaction, | Network construction |
| Drug-disease interactions | N/A | 1,663 (ODV:117) | 466,657 | Drug-Disease Interactions | Network construction |
| GCNMDA [1] | 167( 175-8 no DBID) (ODV:136) | N/A | Drug-virus Prediction methods | State Of The Art (SOTA) | |
| Total Network | (ODV:175) | 487,679 | Heterogeneous Network | Network Construction |
The numbers in the brackets are the original numbers before the data filtration (ODV means the Overlap with DrugVirus datasets, in total, our network has 175 drugs of overlaps).
Pretrained deep-learning models
Here is some information about the pre-trained deep learning models; they are all Tensorflow-based models, without further finetune:
For language models:
Roberta: https://huggingface.co/exbert/?model=xlm-roberta-base
Albert: https://github.com/google-research/albert
Bert: https://huggingface.co/bert-base-uncased
GPT-2: GPT2LMHeadModel
For image-based models, it is also Tensorflow-based, and downloaded from tensorflow.keras.applications, details of the model are as follows:
#model=ResNet101(weights=”imagenet”, include_top=False, pooling=”avg”)
#model=ResNet50(weights=”imagenet”, include_top=False, pooling=”avg”)
#model=EfficientNetV2L(weights=”imagenet”, include_top=False, pooling=”max”)
#model=InceptionResNetV2(weights=”imagenet”, include_top=False, pooling=”avg”)
Different negative sampling strategies
Supplementary Table 3. Details of the constructed datasets
| Constructed datasets | Negative samples from | Selected From No.virus | Selected From No.drug | No.positve samples | No.negative samples | Dataset random seed k |
| Drugvirus (GCNMDA) | GCNMDA drug set | 95 | 175(167 with DBID) | 781 | 781 | 0-4 |
| Drugvirus (same set) | DrugVirus set | 103 | 190 with DBID | 1156 | 1156 | 0-4 |
| Drugvirus (Random Select) | All drugs set | 103 | 9212 with DBID | 1156 | 1156 | 0-4 |
| CTD | All drugs set | 103 | 9212 with DBID | 1592 | 1592 | 0-4 |
We designed transfer validation in the results to reflect the predictive ability of different negative sampling methods. We repeated the sampling process k times (in this manuscript, k=5, we fixed the random seeds to repeat the experiments).
The main difference is that the negative samples are selected from different
The GCNMDA virus/drug features cannot conduct this experiment because of its limited number of viruses/drug entries (samples). Thus, if we use the GCNMDA features for training set construction, the model would have trouble learning the dataset’s distribution. Therefore, we selected a virus and drug embeddings group to conduct the transfer verification.
Part Ⅲ Results
Transfer results
Although for the known pair, there is no overlap, However, as we leveraged the random sampling policy, we cannot guarantee that the negative Overlap does not have overlaps. To further validate if the CTD dataset can be an independent dataset, we further counted the overlaps of our constructed dataset; details are as follow:
| CTD_frs0 | CTD_frs1 | CTD_frs2 | CTD_frs3 | CTD_frs4 | Size | |
| DB_frs0 | 991 | 5 | 3 | 2 | 0 | 2312 |
| DB_frs1 | 5 | 987 | 1 | 3 | 1 | 2312 |
| DB_frs2 | 4 | 3 | 958 | 0 | 1 | 2312 |
| DB_frs3 | 3 | 2 | 0 | 996 | 0 | 2312 |
| DB_frs4 | 1 | 1 | 1 | 1 | 982 | 2312 |
| Size | 3184 | 3184 | 3184 | 3184 | 3184 | – |
Frs_k means the dataset is constructed with the random seed k. As long as it is not using the same random seeds, the Overlap is acceptable.
As can be seen from above, if the CTD and drug virus use the same random seed to make the constructed dataset, it would contain plenty of overlaps. In order to make it fairer to other constructed datasets, thus, the results from the sample random seed would be removed from the results.
The adjusted AUC/AUPR/metric and p-values of transfer results are shown as follows:
P-value_0 means that the P-value of results (Metrics) from the first pair of VDkey: Roberta-ResNet50 in the dataset of DrugVirus compared with other vdkeys. It is the same with _1,_2,_3, meaning Albert-ResNet50,4mer-ResNet50, and 5mer-ResNet50.
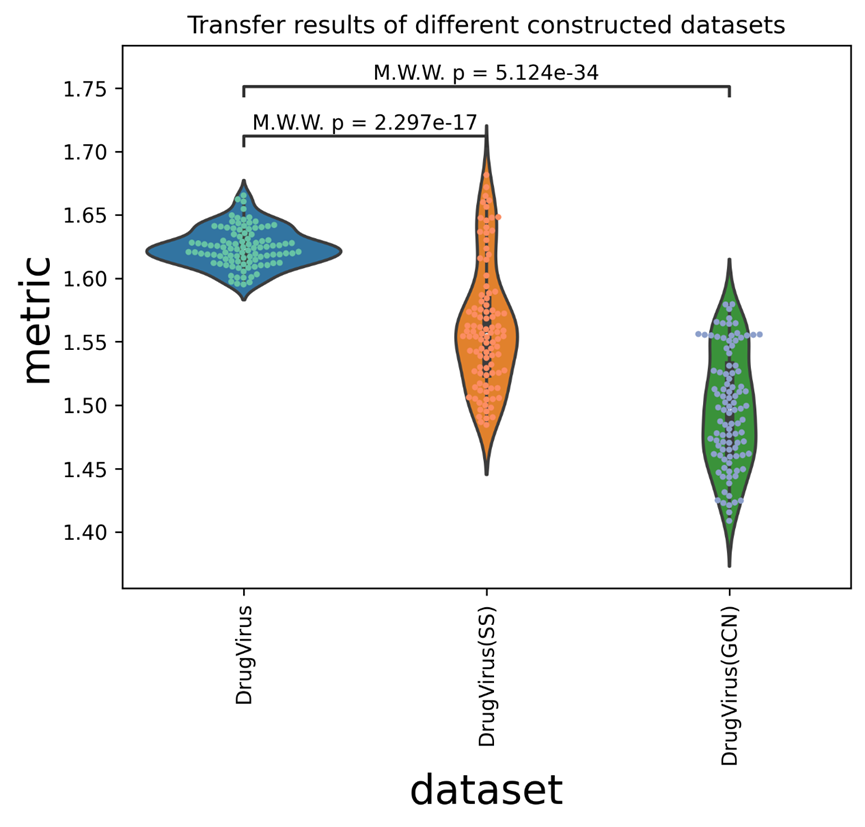
The p-values calculated in the pictures are rank-sum tests with Bonferroni corrections. The results of transfer verification grouped by datasets are shown in Supplementary Figure 1.
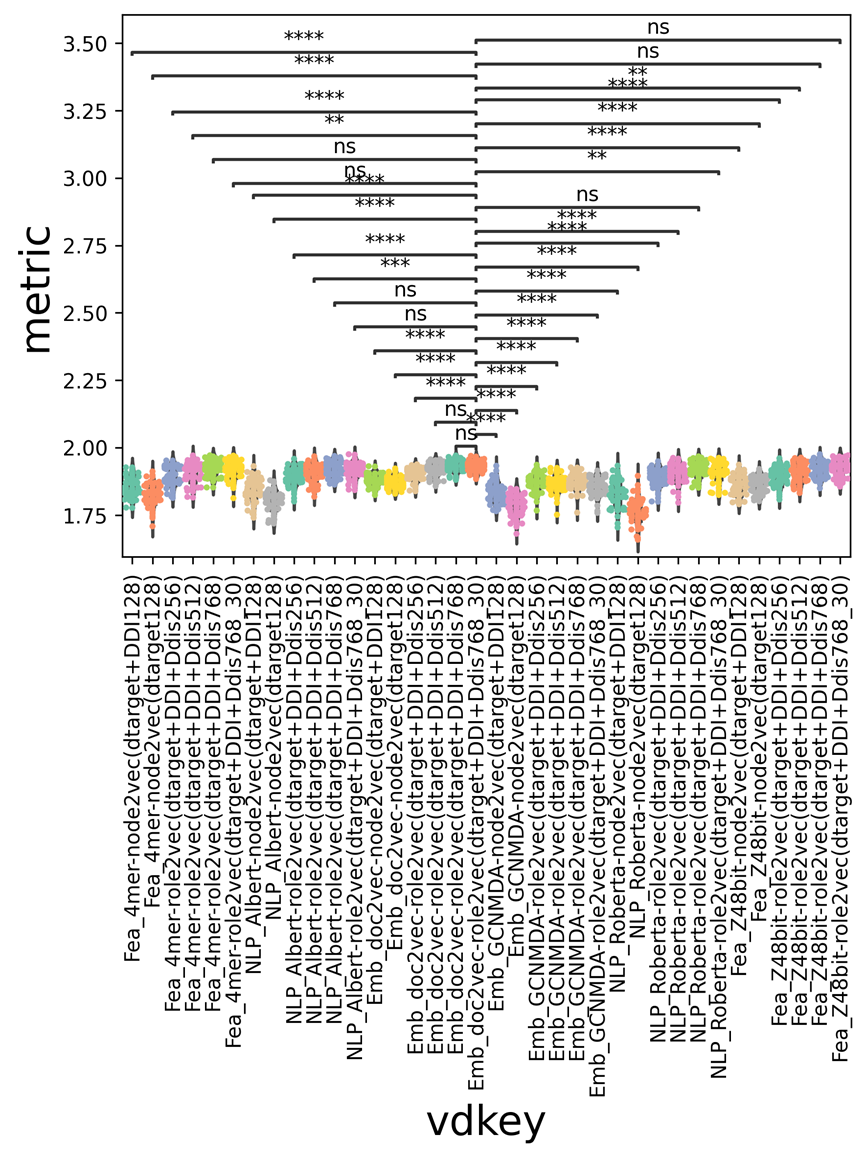
The findings reveal that DrugVirus has considerably superior overall transfer verification outcomes than DrugVirus(SS), with a p-value of 2.297e-17, and DrugVirus(GCN), with a p-value of 5.124e-34.
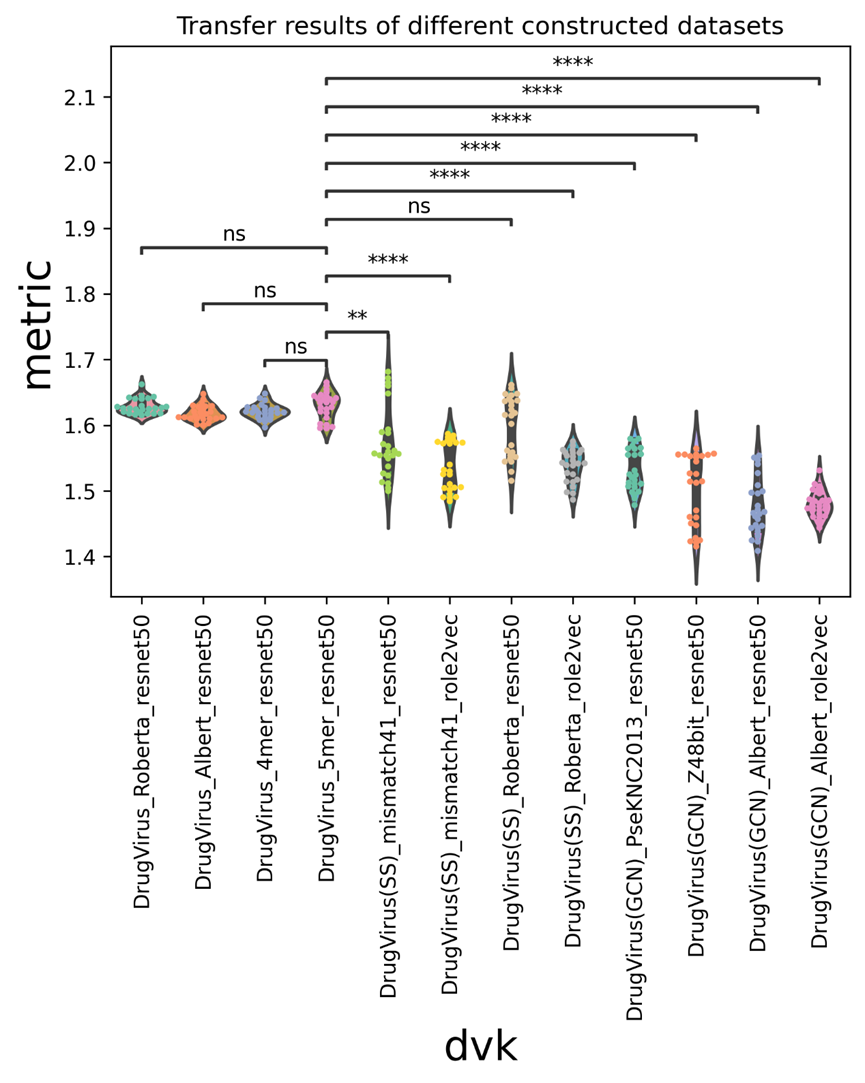
In Supplementary Figure 2, various built datasets’ findings are sorted by vdkeys. We discovered that the 5mer-Resnet vdkey could obtain the greatest average metric (AUC+AUPR). The top four vdkeys of the Drugvirus are distributed similarly to one another in terms of results distribution. However, Drugvirus is not significantly better in the created dataset than Roberta-Resnet (SS). This suggests that compared to other constructed datasets, the Drugvirus-constructed dataset has a greater capacity to predict transfer.
As a result, we carried out more tests, mostly using the Drugvirus dataset that was created.
\
Part Ⅳ Extended Results and Parametrical Analysis
We extracted features from discrepant aspects (Including viruses’ sequential and sematic, drugs’ corpus, Image, and network-based embedding). Some aspects involve parameters, such as virus semantical features and Drug network-based embedding. We conducted some parametrical analysis to filter the best features of those aspects. During this process, we will fix features from other aspects and conduct some experiments on the chosen aspects’ candidate extractors.
Virus Semantical (Corpus-Based)Embedding Finetuning
Due to the limitations of GPU memory, we have to apply different strategies to the original abstracts list. As shown in Supplementary Table 4, we can only generate features from the first ten abstracts if we do not apply any preprocess to the corpus. If we apply a preprocess and a length limitation to the corpus, we could generate features from the first 20 abstracts. Furthermore, if we make the corpus with no overlap words, it will contain information from 100 abstracts. However, this will cause the loss of some information from the frequency of words.
Supplementary Table 4. Details of Finetuning the virus corpus-based embeddings
| Pre-trained-Model | Preprocess Method | 10 Abstracts | 20 Abstracts | 50 Abstracts | 100 Abstracts |
| Albert | Without preprocess | v (able to run) | OOM (Out Of Memory) | OOM | OOM |
| Albert | PreProcess (PP) | v | v | OOM | OOM |
| Albert | PP+Non overlaop (PPNoL) | v | v | v | v |
| Roberta | Without preprocess | v | OOM | OOM | OOM |
| Roberta | PreProcess (PP) | v | v | OOM | OOM |
| Roberta | PP+Non overlaop (PPNoL) | v | v | v | v |
| Bert | Without Preprocess | v | OOM | OOM | OOM |
| Bert | PP | v | v | OOM | OOM |
| Bert | PPNoL | v | v | v | v |
To further explore how those features would affect the performance, we leveraged those features by conducting a parametrical analysis. The dataset we chose is Drugvirus, Type2 verification, and repetition as ten times. The drug virus could perform well on the transfer tasks.
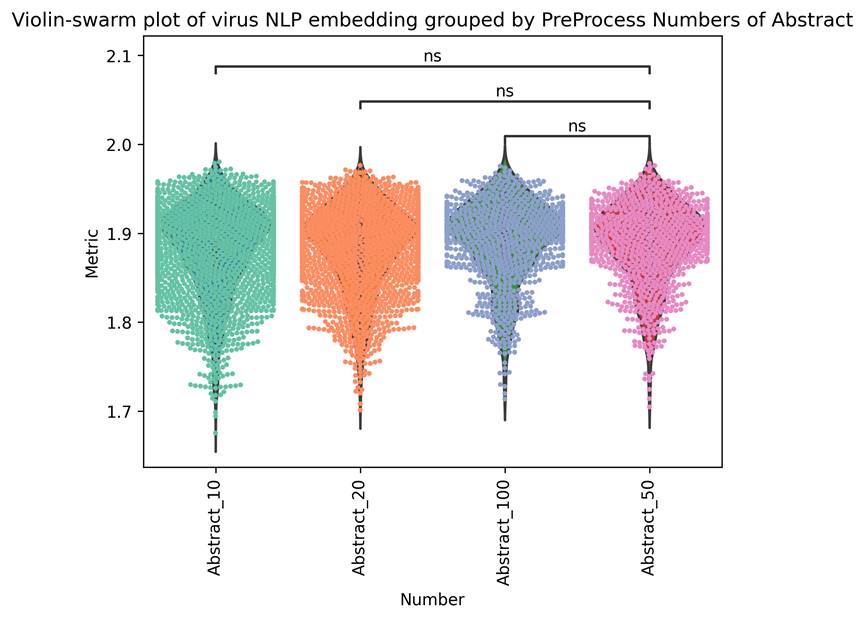
We categorized the parameters and ran various tests. In Supplementary Table 5, the results are displayed. We can see that the improvement is not very noticeable when compared to 10 or 50, or 100 papers (abstract) (Supplementary Figure 3). According to our analysis, each virus’s average abstract number is 86.4606. Although the 100 abstracts are slightly fewer than the 50 abstracts, we still used the 100 abstracts as parameters to create corpus-based embedding in order to add more information.
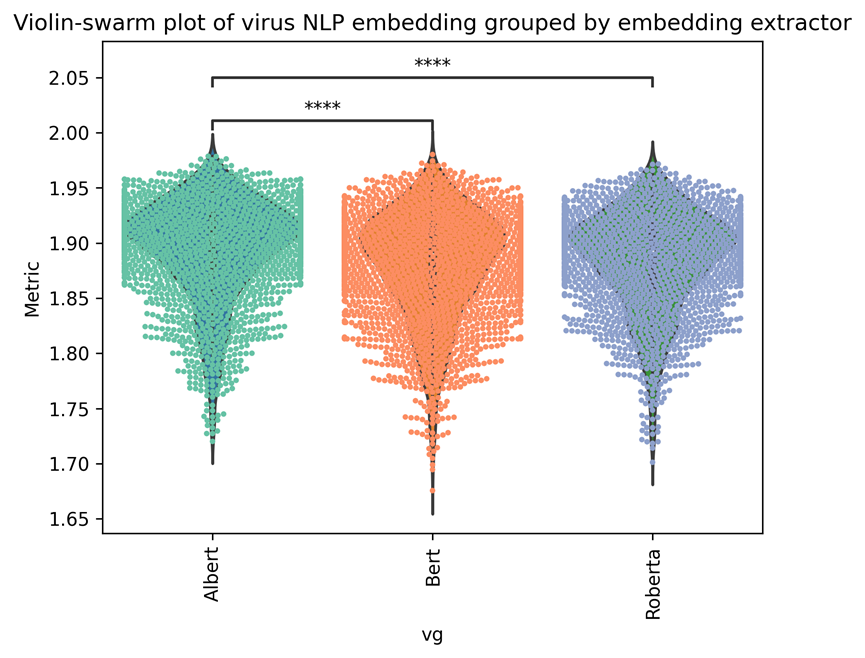
We further sorted the results by using different models to generate context-based embedding. As we can see from Supplementary Figure 4, the Albert is significantly better than other comparison methods with the Rank-sum test with Bonferroni correction (****: p <= 1.00e-04).
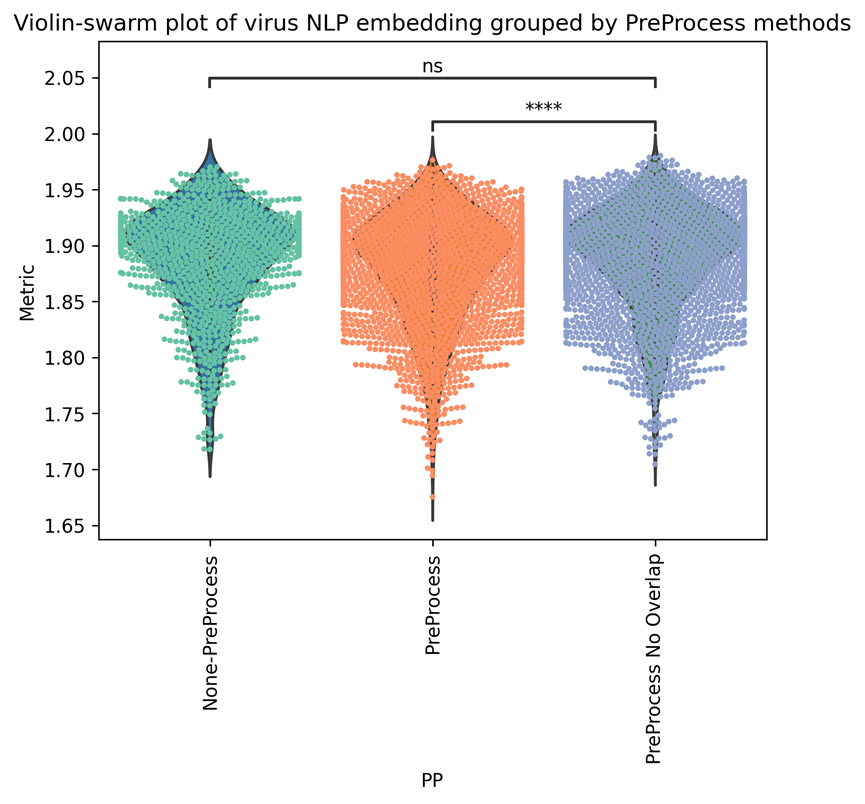
For the methods of preprocessing the corpus, we can see that when no preprocess is employed, the metric is 1.8665; if we applied the PreProcess No Overlap, the average metrics increased to 1.86913. if we only use the Preprocess, it will decrease to 1.8640. it is reasonable that some information might be removed when using the preprocess. However, removing the Overlap can significantly increase performance compared to Preprocessed only, as shown in Supplementary Figure 5. As we want to introduce more semantical information(including more abstracts) into the embedding here, we decide to use the PPNoL as preprocess method to generate corpus-based embeddings.
Thus in the feature pool, we leveraged albertPPNoL100 and robertaPPNoL100 as candidate semantic embedding extractors.
Supplementary Table 5. Results of finetuning the virus corpus-based embeddings
| Numbers of documents | Abstract_10 | Abstract_20 | Abstract_50s | Abstract_100 | figures |
| Metrics (average) | 1.86901 | 1.8640 | 1.86902 | 1.8680 | Figure 3 |
| models | Albert | Bert | Roberta | ||
| Metrics (average) | 1.8764 | 1.8623 | 1.8691 | Figure 4 | |
| Preprocess methods | None-PreProcess | PreProcess | PreProcess No Overlap | ||
| Metrics (average) | 1.8665 | 1.8640 | 1.86913 | Figure 5 |
Drug Network-based Embedding Finetuning
We initially chose no more than two aspects(two sequence features, two sequence embedding, two NLP embedding) for each feature domain of the infection virus and re-paired those features.
Then, in order to build the embedding datasets, we experimented with variously produced network-based drug embeddings and used five-fold cross-validation verification. The table contains information about the parameters used to generate the embedding. Drug network composition (D-target, Drug-Drug, Drug-Disease networks, and their combinations), embedding dimensions, and creation techniques are some of these factors.
In order to create embedding datasets with outstanding transfer verification performance, we used the Drug Virus (random choice for negative samples). Our network-based embedding has 487,697 linkages and 7,592 medicines, whereas the SOTA technique, GCNMDA, has 136 medications. The outcomes are shown in Supplementary Figure 6.
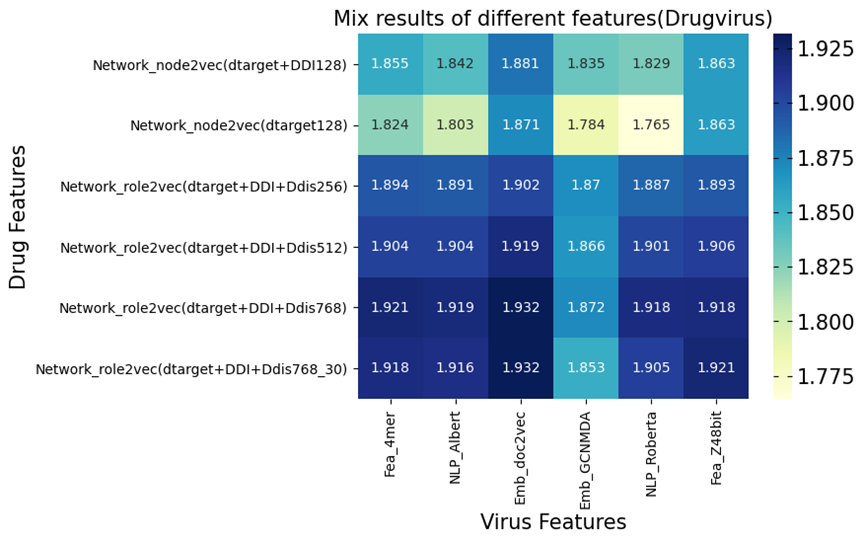
The heatmap demonstrates that performance often rose as the dimension grew. Adding extra links can improve performance, as seen in the first two rows. We further plotted a violin-swam plot with adjusted p-values shown in Supplementary Figure 7,
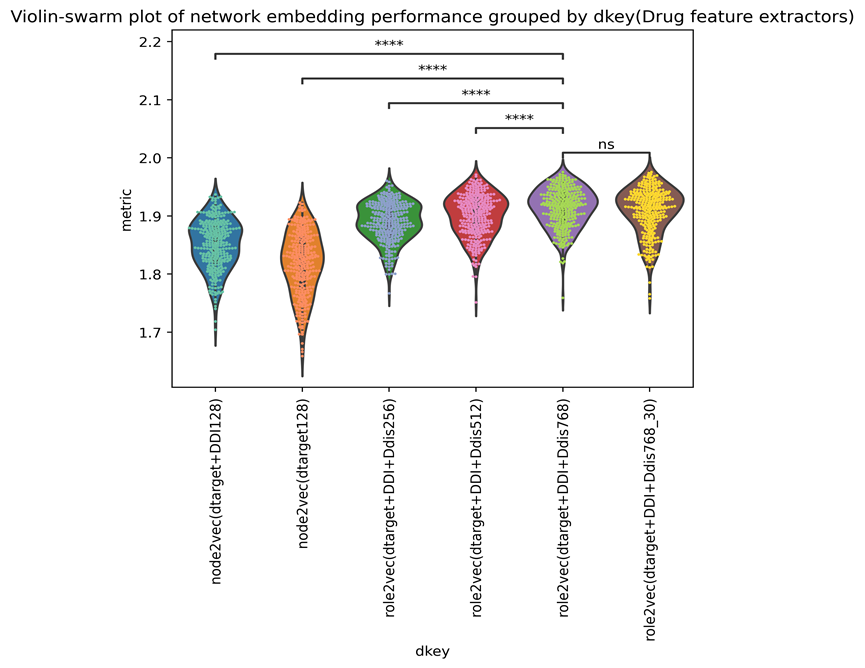
The plot’s trend further suggested that adding dimensions and networks might improve the performance of the model. _30 means that each node will visit 30 times when producing the role2vec embedding. From the figure, it does not improve performance significantly compared with the default setting.
Supplementary Figure 8 shows the output of each virus-drug feature domain extractor (vdkey). We can see that doc2vec-role2vec( Dtarget-DDI-Ddis-768_30) can get the highest mean AUC+AUPR values in this task( with a selected group of virus features).
We ultimately decided to include Drug role2vec(dtarget+DDI+Ddis768 30), and node2vec(dtarget+DDI128) as network representatives to the feature pool for features selection.
Doc2vec finetuning
In our previous work [13], we generated sequence-based embedding for each RNA sequence by first choosing the longest sequence as a sample. Then we generated the embedding for each DNA sequence. We have additional sequences for each virus in this endeavor. Therefore, if a virus has more than one sample, we opted to average the embedding for that virus. In this case, the virus variants are regarded as the same the virus, and their embeddings would be averaged. The longest sequence embedding is marked as doc2vec_longest or doc2vec_(s), and the average embedding is noted as doc2vec_0.8m.
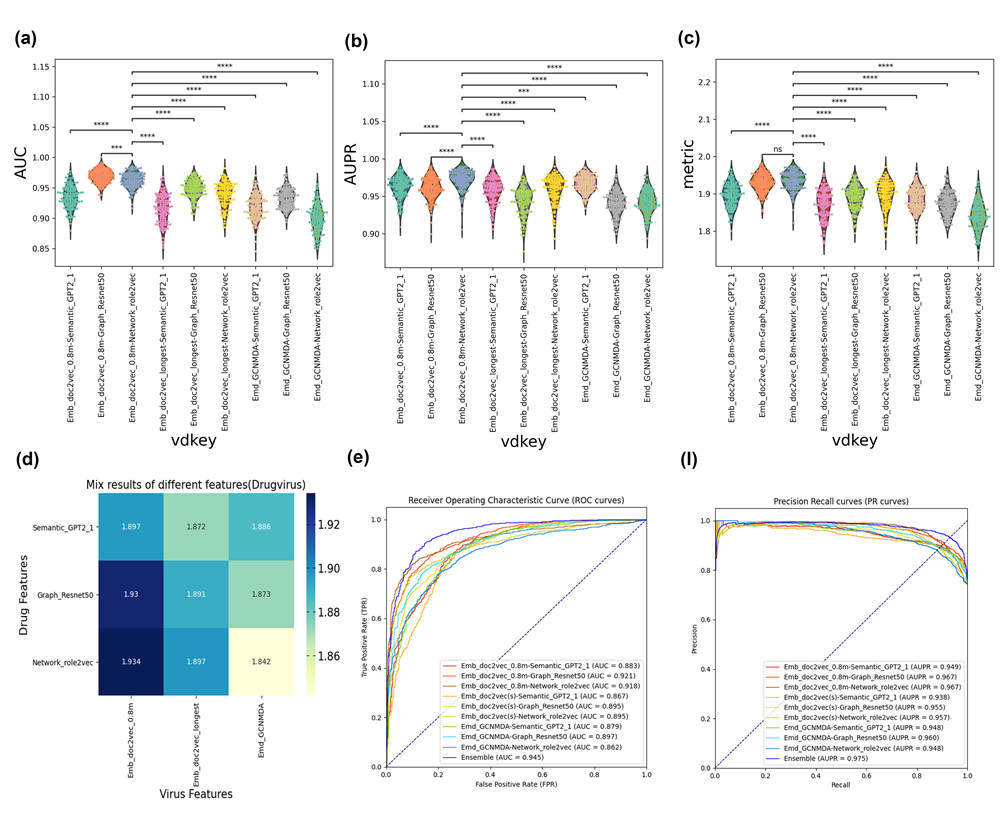
As a SOTA comparison, we also provided the GCNMDA viral features, which are named GCNMDA. We chose one embedding generator for each feature domain for the drug features. The AUC/AUPR/Metric violin-swam plot is shown in Supplementary Figure 9. (a-c). Rank-sum Bonferroni P-values were added at the top of each violin-swam plot. The heatmap of the average metric findings for particular virus-drug feature combinations is shown in Supplementary Figure 9. (d). The ROC/PR curves for those two techniques and GCNMDA are shown in Supplementary Figure 9. (e–f).
The viral averaged embedding doc2vec with drug network embedding role2vec is noticeably superior to the other virus-drug feature domain combinations, as shown in Supplementary Figure 9. (a-c). Additionally, doc2vec_0.8m-role2vec and doc2vec_0.8m-ResNet50 might produce results with a comparable distribution for the overall performance of AUC and AUPR. Supplementary Figure 9. (d). demonstrates that when paired with the three distinct drug feature domains, the viral embedding doc2vec_0.8m can achieve the greatest average metric values.
Part V Case Study of RSV
We further collected respiratory syndrome virus from recent records (December 22, 2022),
We conducted drug repurposing for those newly recorded nucleotide sequences. Results are shown in Supplementary Table 5. As can be seen from the table, DeepSeq2drug performed well, with 10 out of the top 10 predicted medicines being reported from PubMed to treat the contagious RSV. Those PMIDs were also recorded for further research.
The sample IDs are shown in the IDs of the selected sequences for RSV results.
Supplementary Table 5. Predictive results of RSV
| Virus | Drug name | Count | Possible citation from PUBMED |
| respiratory syndrome virus | Ribavirin | 10 | [‘31384456’, ‘33924302’, ‘32352535’, ‘32307245’, ‘27281742’, ‘32634603’, ‘30849247’, ‘33961695’, ‘32282022’, ‘32284326’] |
| respiratory syndrome virus | Chloroquine | 10 | [‘32348588’, ‘32217113’, ‘33010669’, ‘32964796’, ‘32295814’, ‘32446285’, ‘34356617’, ‘33236131’, ‘32696108’, ‘32373993’] |
| respiratory syndrome virus | Mycophenolic acid | 10 | [‘33116299’, ‘32639598’, ‘33743151’, ‘34549821’, ‘33957273’, ‘24323636’, ‘24626235’, ‘25542975’, ‘25810418’, ‘32579258’] |
| respiratory syndrome virus | Amantadine | 10 | [‘31275265’, ‘14643124’, ‘34152583’, ‘1048031’, ‘3500376’, ‘35390511’, ‘33364201’, ‘10965680’, ‘25446940’, ‘15071371’] |
| respiratory syndrome virus | Nitazoxanide | 10 | [‘33336780’, ‘33361100’, ‘32768971’, ‘33588727’, ‘35130104’, ‘27095301’, ‘34755538’, ‘35069994’, ‘28500431’, ‘36094778’] |
| respiratory syndrome virus | Itraconazole | 10 | [‘32428379’, ‘33666253’, ‘35064041’, ‘34984948’, ‘33472466’, ‘35405278’, ‘27895278’, ‘35229317’, ‘36038303’, ‘29899416’] |
| respiratory syndrome virus | Nelfinavir | 10 | [‘33817567’, ‘33482181’, ‘34755538’, ‘16312205’, ‘33217030’, ‘34611467’, ‘33995308’, ‘33080984’, ‘32259313’, ‘32705942’] |
| respiratory syndrome virus | Amodiaquine | 10 | [‘33941899’, ‘34755538’, ‘34239286’, ‘36453012’, ‘34217752’, ‘32916297’, ‘34541995’, ‘17176632’, ‘32805422’, ‘33475021’] |
| respiratory syndrome virus | Sunitinib | 4 | [‘34951532’, ‘32540268’, ‘32669298’, ‘35738348’] |
| respiratory syndrome virus | Gemcitabine | 7 | [‘29795047’, ‘33557278’, ‘33479570’, ‘32563698’, ‘36213871’, ‘36334362’, ‘35971500’] |
IDs of the selected sequences for RSV results
“>OP730529.1 |Porcine reproductive and respiratory syndrome virus strain PRRSV-CH-SDLY27-2022 envelope glycoprotein gene, complete cds
“>OP730530.1 |Porcine reproductive and respiratory syndrome virus strain PRRSV-CH-SDLY28-2022 envelope glycoprotein gene, complete cds
“>OP730531.1 |Porcine reproductive and respiratory syndrome virus strain PRRSV-CH-SDLY32-2022 envelope glycoprotein gene, complete cds
“>OP785693.1 |Porcine reproductive and respiratory syndrome virus isolate PRRSV-CH-SDLY176-2022 nonstructural protein 2 gene, partial cds
“>OP785694.1 |Porcine reproductive and respiratory syndrome virus isolate PRRSV-CH-SDLY177-2022 nonstructural protein 2 gene, partial cds
“>OP785695.1 |Porcine reproductive and respiratory syndrome virus isolate PRRSV-CH-SDLY28-2022 nonstructural protein 2 gene, partial cds
“>OP785696.1 |Porcine reproductive and respiratory syndrome virus isolate PRRSV-CH-SDLY32-2022 nonstructural protein 2 gene, partial cds
“>OM677752.1 |Porcine reproductive and respiratory syndrome virus strain NPUST3064 glycoprotein 5 gene, complete cds
“>OM677753.1 |Porcine reproductive and respiratory syndrome virus strain 108-355 glycoprotein 5 gene, complete cds
“>OM677754.1 |Porcine reproductive and respiratory syndrome virus strain 108-603 glycoprotein 5 gene, complete cds
“>OM677755.1 |Porcine reproductive and respiratory syndrome virus strain NPUST3554 glycoprotein 5 gene, complete cds
“>OM677756.1 |Porcine reproductive and respiratory syndrome virus strain NPUST3599 glycoprotein 5 gene, complete cds
“>OM677757.1 |Porcine reproductive and respiratory syndrome virus strain 108-2275 glycoprotein 5 gene, complete cds
“>OM677758.1 |Porcine reproductive and respiratory syndrome virus strain NPUST4028 glycoprotein 5 gene, complete cds
“>OM677759.1 |Porcine reproductive and respiratory syndrome virus strain NPUST4035 glycoprotein 5 gene, complete cds
“>OM677760.1 |Porcine reproductive and respiratory syndrome virus strain NPUST4178 glycoprotein 5 gene, complete cds
“>OM677761.1 |Porcine reproductive and respiratory syndrome virus strain NPUST4260 glycoprotein 5 gene, complete cds
“>OM677762.1 |Porcine reproductive and respiratory syndrome virus strain 109-920 glycoprotein 5 gene, complete cds
“>OM686875.1 |Porcine reproductive and respiratory syndrome virus strain 103-555 ORF5 gene, complete cds
“>OM801587.1 |Porcine reproductive and respiratory syndrome virus strain KN-21035TB envelope protein GP5, membrane protein GP6, and nucleocapsid protein genes, complete cds
“>OM860456.1 |Porcine reproductive and respiratory syndrome virus isolate VNUA-PRRS-HY-01 glycoprotein 5 (GP5) gene, complete cds
“>MK279739.1 |Porcine reproductive and respiratory syndrome virus strain PRRSV/pig/CHN/JTS/201606, complete genome
“>MK279740.1 |Porcine reproductive and respiratory syndrome virus strain PRRSV/pig/CHN/TG/201711, complete genome
“>MK279741.1 |Porcine reproductive and respiratory syndrome virus strain PRRSV/pig/CHN/JK/201805, complete genome
“>ON691479.1 |Porcine reproductive and respiratory syndrome virus strain GD-H1, complete sequence
“>ON691480.1 |Porcine reproductive and respiratory syndrome virus strain GD-H1, complete sequence
Supplementary Table 6.
DeepSeq2drug Top Predicted Drug for Monkeypox Virus
(Ref seq and average seq)
| Rank | Drug_name (Ref_seq) | Count | REF | Drug_name (200_Monkeypox) | REF |
| 1 | Ribavirin | 2 | [51], [52] | Ribavirin | [51], [52] |
| 2 | Amantadine | 0 | Chloroquine | 0 | |
| 3 | Mycophenolic acid | 1 | [51] | Mycophenolic acid | [51] |
| 4 | Chloroquine | 0 | Nitazoxanide | 0 | |
| 5 | Itraconazole | 0 | Itraconazole | 0 | |
| 6 | Nitazoxanide | 0 | Amantadine | 0 | |
| 7 | Nelfinavir | 0 | Nelfinavir | 0 | |
| 8 | Amodiaquine | 0 | Amodiaquine | 0 | |
| 9 | Sorafenib | 0 | Sunitinib | 0 | |
| 10 | Gemcitabine | 1 | [53] | Sorafenib | 0 |
| 11 | Sunitinib | 0 | Gemcitabine | [53] |
The Monkeypox ref_seq (NCBI Accession ID GCF_000857045.1) and 200 monkeypox DNA nucleotide sequences (NCBI Accession ID range from ON669283.1 to OP881933.1, searched on 12th Dec 2022) are the parameters that control the embedding is generated by one sequence or a batch of sequences. We can find out that the predictive results of the Reference sequence and a batch of recent Monkeypox are slightly different.
Reference
- [2] Chen Z, Zhao P, Li F, et al. iLearn: an integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data[J]. Briefings in bioinformatics, 2020, 21(3): 1047-1057.
- [4] Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, Assempour N, Iynkkaran I, Liu Y, Maciejewski A, Gale N, Wilson A, Chin L, Cummings R, Le D, Pon A, Knox C, Wilson M. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2017 November 8. doi: 10.1093/nar/gkx1037.